以下のように、DNAに基づいて個人を識別するプログラムを実装します。
$ python dna.py databases/large.csv sequences/5.txt
Lavender
始め方
VS Codeを開きます。
ターミナルウィンドウ内をクリックすることから始めて、それからcdを実行します。
その後プロンプトは次のようになっていることがわかります。
$ターミナルウィンドウの内側をクリックし、次のように入力します。
wget https://cdn.cs50.net/2021/fall/psets/6/dna.zipその後にEnterを押すと、dna.zipというZIPがあなたのCodespaceにダウンロードされます。wgetと次のURLの間にあるスペースや、その他の文字を見落とさないように注意してください。
次に
unzip dna.zipを実行して、dnaというフォルダを作成します。
ZIPファイルは不要になったため、
rm dna.zipを実行し、プロンプトで “y “に続いてEnterで応答すると、ダウンロードしたZIPファイルが削除されます。
次に
cd dnaの後にEnterを押して、そのディレクトリに移動する(つまり、開く)。これでプロンプトは以下のようになります。
dna/ $lsを実行するいくつかのファイルとフォルダーが表示されます。
databases/ dna.py sequences/問題が発生した場合は、同じ手順をもう一度実行して、どこが間違っていたかを判断できるかどうかを確認してください。
背景
生物の遺伝情報を担うDNAは、数十年にわたって刑事司法で利用されてきました。ですが、DNAプロファイリングは正確にはどのように機能するのでしょうか。DNAの塩基配列が与えられたとき、法医学者はどのようにしてそれが誰に属するかを同定できるのでしょうか。
DNAはヌクレオチドと呼ばれる分子の配列で、特定の形 (2重らせん) に配列しています。ヒトのすべての細胞には数10億個のヌクレオチドが配列しています。DNAの各ヌクレオチドは、アデニン (A) 、シトシン (C) 、グアニン (G) 、チミン (T) の4種類の塩基のうちの1つを含んでいます。この配列のいくつかの部分 (ゲノム) は、ほぼすべてのヒトで同一であるか、または少なくとも非常に類似していますが、配列の他の部分はより高い遺伝的多様性を有し、したがって集団全体ではより多様なものとなります。
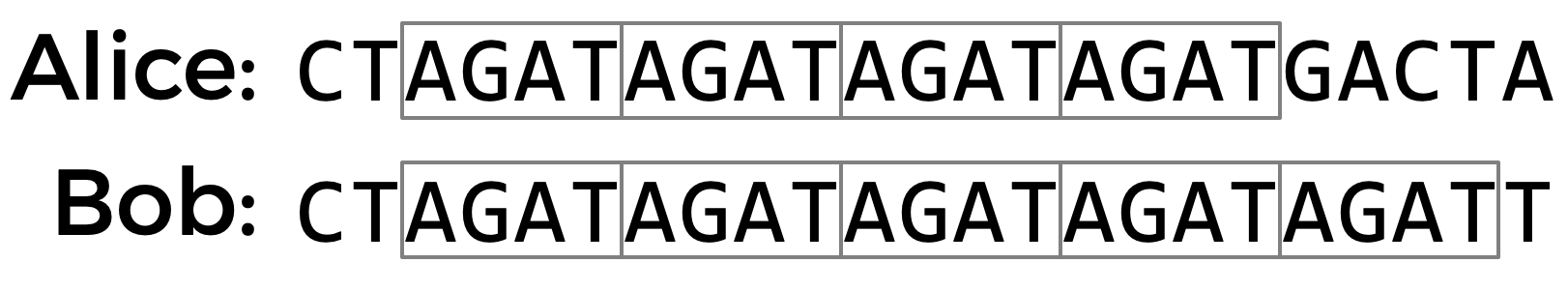
DNAが高い遺伝的多様性を有する傾向がある1つの場所は、Short Tandem Repeats (STR) です。STRはDNA塩基の短い配列で、DNAの特定の位置で連続的に何度も繰り返される傾向があり、個々のSTR反復回数は個体によって大きく異なります。例えば、以下のDNAサンプルでは、AliceのDNAでSTR AGATが4回繰り返されており、Bobは同じSTRが5回繰り返されています。
1つではなく複数のSTRを使用することで、DNAプロファイリングの精度を向上させることができます。2人がひとつのSTRについて同じ数の繰返しをもつ確率が5%であり、解析者が10の異なるSTRに注目した場合、2つのDNA試料が偶然に一致する確率は約1000兆分の1です (すべてのSTRが互いに独立していると仮定すします) 。したがって、もし2つのDNAサンプルがそれぞれのSTRで繰り返し一致すれば、解析者はそれらが同一人物からのものであるとかなり確信することができます。FBIのDNAデータベースであるCODISは、DNAプロファイリングプロセスの一環として20種類のSTRを使用しています。
このようなDNAデータベースはどのようなものでしょうか。最も単純な形式では、DNAデータベースをCSVファイルとしてフォーマットすることが考えられます。CSVファイルでは、各行は個人に対応し、各列は特定のSTRに対応します。
name,AGAT,AATG,TATC
Alice,28,42,14
Bob,17,22,19
Charlie,36,18,25
上記のファイルのデータから、アリスはAGATという配列をDNAのどこかで28回連続して繰り返し、AATGという配列を42回繰り返し、TATCという配列を14回繰り返していることがわかります。一方、Bobは同じ3つのSTRをそれぞれ17回、22回、19回繰り返しています。チャーリーは同じ三つのSTRをそれぞれ36回、18回、25回繰り返しています。
DNAの塩基配列からそれが誰のものかどうやって特定するのでしょう?あなたがDNA配列を調べて、反復されるAGATの最も長い連続した配列を探し、最も長い配列が17回の長さであることを見つけたと想像してみてください。もしAATGの最も長い配列が22回の繰り返しであり、TATCの最も長い配列が19回の繰り返しであることがわかれば、DNAがボブのものであるというかなりよい証拠を提供することになります。もちろん、各STRのカウントを取得すると、DNAデータベース内の誰にも一致しなくなる可能性もあります。この場合、一致なしということになります。
実際には、解析者はどの染色体上のどの位置にSTRが見つかるかを知っているので、DNAのごく一部に検索を限定することができますが、この問題ではその詳細は無視しましょう。
あなたの課題は、DNAのシークエンスと個人のリストのSTR反復回数を含むCSVファイルを取るプログラムを書くことです。そして、DNAが (おそらく) 誰に属するかを出力します。
仕様
dna.pyというファイルに、DNAのシークエンスが誰に属するかを識別するプログラムを実装します。
- プログラムの最初のコマンドライン引数として、個人のリストのSTR反復回数を含むCSVファイルの名前を必要とし、2番目のコマンドライン引数として、識別するDNA配列を含むテキストファイルの名前を必要とします。
- 誤った数のコマンドライン引数を指定してプログラムを実行すると、プログラムは選択したエラーメッセージを (
printを使用して) 出力します。引数の数が正しい場合は、最初の引数が実際に有効なCSVファイルのファイル名であり、2番目の引数が有効なテキストファイルのファイル名であると仮定できます。
- 誤った数のコマンドライン引数を指定してプログラムを実行すると、プログラムは選択したエラーメッセージを (
- プログラムはCSVファイルを開き、その内容をメモリに読み込みます。
- CSVファイルの最初の行が列名であると仮定します。最初の列は単語名で、残りの列はSTRシーケンスそのものです。
- プログラムはDNA配列を開き、その内容をメモリに読み込む必要があります。
- 各STR (CSVファイルの最初の行から) について、プログラムは同定するためにDNA配列中のSTRの連続反復の最長の数を計算しなければなりません。ヘルパー関数
longest_matchが定義されていることに注目してください。 - STR反復回数がCSVファイル内の個人のいずれかと完全に一致する場合、プログラムは一致する個人の名前を出力する必要があります。
- STR反復回数は、複数の個人に一致しないと想定できます。
- 反復回数がCSVファイルのどの個人とも正確に一致しない場合、プログラムは
"No match"を出力します。
ウォークスルー
使い方
プログラムは次の例のように動作します。
$ python dna.py databases/large.csv sequences/5.txt
Lavender
$ python dna.py
Usage: python dna.py data.csv sequence.txt
$ python dna.py data.csv
Usage: python dna.py data.csv sequence.txtヒント
- Pythonの
csvモジュールは、CSVファイルをメモリに読み込むのに便利です。csv.readerまたはcsv.DictReaderのいずれかを利用できます。 open関数とread関数は、テキストファイルをメモリに読み込む場合に便利です。- プログラム内の情報を追跡するのに役立つデータ構造を検討します。
listやdictが役に立つかもしれません。 - DNA配列とSTRを入力とし、STRの最大繰り返し回数を返す関数(
longest_match)を定義したことを思い出してください。この関数はプログラムの他の部分で使用することができます。
テスト
この問題についてはcheck50 を使用できますが、次の各項目については、最初に自分でコードをテストすることをお勧めします。
- プログラムを
python dna.py databases/small.csv sequences/1.txtとして実行します。プログラムはBobを出力するはずです。 - プログラムを
python dna.py databases/small.csv sequences/2.txtとして実行します。プログラムはNo matchと出力するはずです。 - プログラムを
python dna.py databases/small.csv sequences/3.txtとして実行します。プログラムはNo matchと出力するはずです。 - プログラムを
python dna.py databases/small.csv sequences/4.txtとして実行します。プログラムはAliceを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/5.txtとして実行します。プログラムはLavenderを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/6.txtとして実行します。プログラムはLunaを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/7.txtとして実行します。プログラムはRonを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/8.txtとして実行します。プログラムはGinnyを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/9.txtとして実行します。プログラムはDracoを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/10.txtとして実行します。プログラムはAlbusを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/11.txtとして実行します。プログラムはHermioneを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/12.txtとして実行します。プログラムはLilyを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/13.txtとして実行します。プログラムはNo matchと出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/14.txtとして実行します。プログラムはSeverusを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/15.txtとして実行します。プログラムはSiriusを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/16.txtとして実行します。プログラムはNo matchと出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/17.txtとして実行します。プログラムからHarryが出力されます。 - プログラムを
python dna.py databases/large.csv sequences/18.txtとして実行します。プログラムはNo matchと出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/19.txtとして実行します。プログラムはFredを出力するはずです。 - プログラムを
python dna.py databases/large.csv sequences/20.txtとして実行します。プログラムはNo matchと出力するはずです。
check50を使用して以下を実行し、コードの正確さを評価してください。ただし、テストは必ず自分で行ってください。
check50 cs50/problems/2022/x/dna以下を実行し、style50を使用してコードのスタイルを評価します。
style50 dna.py提出方法
ターミナルで、以下のコマンドを実行して提出してください。
submit50 cs50/problems/2022/x/dna